Gruppo IMER - Regione Emilia Romagna - NewsLetter 25
Atresia esofagea
Registro IMER (1981-2000)
Rivieri F., Miolo GM., Astolfi G., Palazzi P., Neville AJ., Calzolari E.
L’atresia esofagea è una discontinuità congenita del lume dell’esofago in cui la porzione prossimale e distale dell’esofago terminano a fondo cieco. Un moncone a fondo cieco dell’esofago, più spesso quello distale, può comunicare, attraverso una fistola (detta tracheoesofagea), con la trachea.
Tipi di atresia esofagea
Sono stati descritti 5 tipi anatomici di atresia esofagea: tipo A) atresia esofagea senza fistola; tipo B) atresia esofagea con fistola che connette il moncone superiore con la trachea; tipo C) atresia esofagea con fistola che connette il moncone inferiore con la trachea; tipo D) atresia esofagea con fistola sia nel moncone superiore che nel moncone inferiore che connettono con la trachea; tipo E) fistola tracheoesofagea senza atresia esofagea (fistola ad H).
Embiologia
L’esofago e la trachea derivano dall’intestino cefalico primitivo. Durante la 4a settimana di sviluppo embrionale, la trachea si forma come un diverticolo ventrale dalla faringe primitiva (parte caudale dell’intestino cefalico primitivo). Un setto tracheoesofageo si sviluppa ove le pieghe tracheoesofagee longitudinali si fondono insieme. Questo setto divide, nel corso della 5a settimana di sviluppo, l’intestino primitivo in una porzione ventrale, il tubo laringotracheale, e una porzione dorsale, l’esofago.
L’atresia esofagea risulta se il setto tracheoesofageo è deviato posteriormente, causando una incompleta separazione dell’esofago dal tubo laringotracheale e dando origine ad una concomitante fistola tracheoesofagea.
L’atresia esofagea senza fistola, viene attribuita ad un fallimento della ricanalizzazione dell’esofago durante 8a settimana di sviluppo embrionale.
Riguardo alla embriogenesi dell’atresia esofagea, con o senza fistola, sono state proposte alcune teorie, tra le quali insufficienza vascolare, non sincronia della crescita del mesenchima e dell’epitelio esofageo, anomalie della notocorda, coinvolgimento delle cellule della cresta neurale (Possoegel et al. 1999; Otten et al. 2000; Oxford et al. 2000; Konstantinidou et al. 2001; Morini et al. 2001).
Eziologia
L’eziologia dell’atresia esofagea e della fistola tracheoesofagea non è attualmente nota.
Sebbene in letteratura siano stati descritti alcuni casi familiari, i gemelli monozigoti non sono generalmente concordanti per tale anomalia, e il modello di ereditarietà mendeliano non è probabile (Torfs et al. 1995).
Gli studi che hanno valutato la presenza di atresia esofagea tra i familiari dei casi indice, riportano una bassa ricorrenza nei fratelli nati precedentemente e nei figli dei casi indice (David et al. 1974, 1975; Warren et al. 1979; Staey et al. 1984; Szendrey et al. 1985).
Diversi studi riportano una alta incidenza di gemellarità tra i casi di atresia esofagea, suggerendo che la gemellarità stessa possa essere un fattore predisponente di atresia esofagea, comportando una maggior suscettibilità a teratogeni e/o a carenze nutrizionali/vascolari (David et al. 1974, 1975; Depaepe et al. 1993; Robert et al. 1993; Torfs et al. 1995, Harris et al. 1995; Oxford et al. 2000).
Questi dati nel loro insieme suggeriscono l’importanza di fattori non genetici nell’occorrenza di tale anomalia.
In passato è stata riportata una variabilità stagionale da alcuni studi (David et al. 1974; Squire E.N. 1977) che ha fatto supporre una eziologia infettiva, tuttavia non confermata (Fraser et al. 1987).
Esposizione a farmaci quali ormoni femminili esogeni sono stati suggeriti quali possibili fattori eziologici (Nora et al. 1975), seppur non confermati da altri studi (Torfs et al. 1981; Lammer et al. 1986).
Sono stati descritti alcuni case-report di nati da madri ipertiroidee, che avevano assunto metimazolo in gravidanza, i quali presentavano atresia esofagea e fistola tracheoesofagea (Ramirez et al 1992; Di Giannantonio et al 2001). Gli autori ipotizzano che l’atresia esofagea potrebbe essere una anomalia specifica dell’embriopatia da metimazolo; tuttavia un recente studio (Seoud et al. 2003) pone il problema se sia l’ipertiroidismo non trattato, e non l’assunzione di metimazolo, ad essere teratogeno.
Deficit vitaminici, in particolare della vitamina A, sono stati riportati quali fattori di rischio (Torfs, abstract on line).
Prevalenza
La prevalenza media dell’atresia esofagea, nel periodo 1992-1999, è di circa 2,6 (l.c. 2,3-2,8) per 10000 nati in Italia (rapporto ISTISAN 2002; Tabella 1).
In Europa la prevalenza media dell’atresia esofagea è di circa 2,9 per 10000 nati, con un range di 1,9 - 7,9 per 10000 nati (dati EUROCAT 1980-1999, report 8, 2002).
Studi epidemiologici riportano una ampia variabilità della prevalenza alla nascita tra i differenti registri internazionali di malformazioni congenite (da 0,4 a 3,6 per 10.000 nati vivi; Robert et al 1993).
Anomalie associate
Vari studi riportano che circa il 32-58% dei casi con atresia esofagea presentano anomalie associate (David et al. 1975, Chittmittrapap et al. 1989, Depaepe et al. 1993, Robert et al. 1993; Torfs et al. 1995; Harris et al. 1995; Brown et al 1999, Sparey et al. 2000, Heurn et al. 2002).
Le anomalie del sistema cardiovascolare (19-49%) e gastrointestinali (circa 11-22 % dei casi), in particolare anorettali, sono le più frequentemente associate (Chittmittrapap et al. 1989, Sparey et al. 2000, Heurn et al. 2002).
L’associazione di VATER (anomalie Vertebrali, atresia Anale, TracheoEsofagee, Renali e del Radio) o la più estesa VACTERL (con anomalie Cardiovascolari e degli arti) è una delle condizioni note più frequentemente riscontrate.
E’ riportata una associazione tra atresia esofagea e l’arteria ombelicale unica (la quale, a sua volta, può essere associata a difetti renali silenti) (Torfs et al. 1995).
Le condizioni cromosomiche più frequentemente riportate nei casi con atresia esofagea sono la trisomia 21 e 18 (Depaepe et al. 1993; Robert et al. 1993; Torfs et al. 1995; Harris et al. 1995).
Diversi studi riportano come la presenza di altre anomalie associate, in relazione al numero e al grado di severità di queste (soprattutto se cardiovascolari), siano associate ad una minore sopravvivenza dei nati (David et al. 1975; Chittmittrapap et al. 1989; Robert et al. 1993; Sparey et al. 2000).
Sex ratio
Diversi studi riportano una predominanza del sesso maschile nei casi di atresia esofagea rispetto alla popolazione generale, in particolare nei casi con anomalie associate (esclusi i casi cromosomici ove invece si osserva una predominanza del sesso femminile, soprattutto per i casi di trisomia 18) (David et al. 1974, 1975; Depaepe et al. 1993; Robert et al. 1993; Harris et al. 1995; Torfs et al. 1995).
Gemellarità
E’ stato osservato un incremento di gemellarità di circa 2-3 volte rispetto alla popolazione generale, soprattutto nei casi con anomalie associate (David et al. 1974, 1975; Depaepe et al. 1993; Robert et al. 1993; Harris et al. 1995; Torfs et al. 1995; Oxford et al. 2000,).
Basso peso alla nascita e prematurità
Molti studi riportano un maggior numero di basso peso alla nascita (1/3 pesa meno di 2250 g) e di nati prematuri rispetto ai controlli, soprattutto tra i casi con altre anomalie associate (David et al. 1975; Depaepe et al. 1993; Robert et al. 1993; Torfs et al. 1995).
Etnicità
Alcuni studi hanno evidenziato una più alta incidenza di atresia esofagea nelle popolazioni bianche rispetto a quelle non bianche (Torfs et al. 1995; Heurn et al. 2002).
Età materna e parità
Una correlazione tra rischio di atresia esofagea ed età materna è stata riportata in alcuni studi, mostrando una incidenza maggiore in donne con età inferiore ai 20 anni e con età pari o superiore a 35 anni (David et al 1975; Szendrey et al 1985). Altri studi evidenziano un incremento di rischio con l’aumentare dell’età materna (Harris et al 1995; Sipek et al. 2002).
Riguardo alla parità viene riportato da alcuni studi un incremento di rischio in donne con parità 1 (stratificato per età materna) suggerendo una possibile associazione a problemi di infertilità (Roberts et al. 1993; Harris et al. 1995).
Diagnosi prenatale
L’atresia esofagea viene diagnosticata prenatalmente, secondo studi recenti, in circa 8,9-24% dei casi.
Il riscontro della combinazione di polidramnios (generalmente riscontrato nel corso del III° trimestre) e di assenza della bolla gastrica ha un valore predittivo positivo di atresia esofagea del 56%. Il riscontro della tasca a fondo cieco a livello del collo fetale, seppur non evidente prima della 26° settimana di gestazione, è un reperto ecografico che sembra avere un’alta specificità per l’atresia esofagea (Sparey et al. 2000).
Casistica IMER (1981-2000)
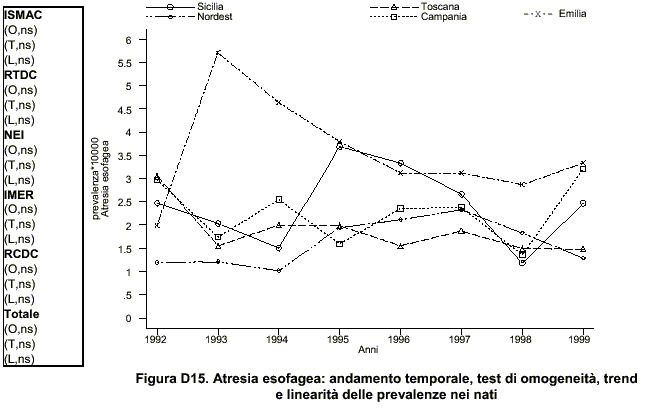
Prevalenza e trend temporale
172 casi (nati vivi, nati morti e interruzioni terapeutiche di gravidanza) di atresia esofagea, con o senza fistola tracheoesofagea (codice ICD9 750300-750310), sono stati accertati nel periodo 1981-2000 nel registro IMER tra 471517 nati sorvegliati.
La prevalenza alla nascita risulta di 3,6 (l.c. 3,1-4,2) per 10000 nati e rientra nell’intervallo riportato dai dati di letteratura.
I nati vivi costituiscono la maggioranza dei casi (170/172=98,9%). Due casi sono costituiti da un nato morto e una interruzione terapeutica di gravidanza a seguito di diagnosi prenatale.
Nel periodo di tempo in esame non sono state osservate variazioni significative nella prevalenza sia per le forme isolate che per le forme associate (tabella 2).
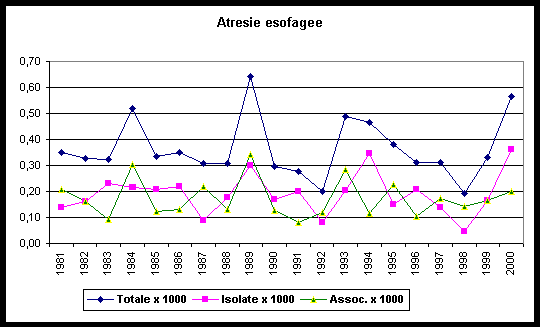 Tabella 2.Andamento temporale (1981-2000) della prevalenza alla nascita dell’atresia esofagea
Tabella 2.Andamento temporale (1981-2000) della prevalenza alla nascita dell’atresia esofagea
P=0,63 (N.S.)
Casi isolati e associati
I casi sono stati suddivisi in isolati (91; 52,9%) e associati (81; 47,1%); questi ultimi ripartiti in anomalie multiple (MCA), condizioni note (sindromi, associazioni, sequenze) e cromosomiche (tabella 3). La distribuzione tra i casi isolati e associati è in accordo con la letteratura.
|
Tabella 3: Atresie esofagee con o senza fistola |
|||||
|
Isolate |
Associate |
Totale |
|||
|
Tipo di atresia |
|
MCA |
Condizioni note |
Cromosomiche |
|
|
Atresia esofagea senza fistola |
40 |
22 |
4 |
6 |
72 |
|
Atresia esofagea con fistola |
51 |
28 |
15 |
6 |
100 |
|
Totale |
91 |
50 |
19 |
12 |
172 |
Tra i 91 casi isolati, 40 sono atresie esofageee senza fistola (44%) e 51 sono associate a fistola tracheoesofagea (56%).
I casi di MCA sono 50 (29,1% sul totale; 62% dei casi associati) di cui 22 casi di di atresia esofagea senza fistola (44%) e 28 casi di atresia esofagea con fistola (56%).
Un caso di MCA è rappresentato da una interruzione terapeutica di gravidanza (alla 22° settimana di gestazione) che presentava atresia esofagea con fistola, displasia polmonare e displasia renale .
Le malformazioni associate dei casi di MCA sono rappresentate prevalentemente da anomalie del sistema cardiocircolatorio (48,5 % dei casi), dell’apparato genitourinario (30%) e dell’apparato gastrointestinale (atresie/stenosi anorettali/intestinali) (18%).
Le anomalie del sistema cardiocircolatorio sono più frequenti nei casi di atresia esofagea con fistola (53,5% vs 41%) (tabella 4).
Tabella 4: Numero di malformazioni associate per apparato nei 50 casi con anomalie multiple (MCA)
|
Difetti |
Atresia esofagea senza fistola |
Atresia esofagea con fistola |
Totale |
|
SNC |
4 |
1 |
5 |
|
Occhio |
0 |
2 |
2 |
|
Orecchio-Faccia-Collo |
2 |
3 |
5 |
|
Cardiovascolare |
9 |
15 |
24 |
|
Sistema respiratorio |
0 |
2 |
2 |
|
Labio/palatoschisi |
2 |
1 |
3 |
|
Sistema gastroenterico* |
4 |
5 |
9 |
|
Sistema genito-urinario |
8 |
7 |
15 |
|
Muscoloscheletrico |
4 |
5 |
9 |
|
Arti |
1 |
3 |
4 |
|
Anomalie parete addominale |
0 |
2 |
2 |
|
Totale |
34 |
46 |
80 |
*Esclusa l’atresia esofagea
Le condizioni note sono 19 (11% sul totale, 23,4% dei casi associati) di cui 15 (78,9%) atresia esofagea con fistola.
Tra le condizioni note vi sono: 15 casi di VATER (delle quali 12 con atresia esofagea associata a fistola), 2 CHARGE (entrambe con atresia esofagea associata a fistola), 1 caso di toxoplasmosi congenita e 1 caso di sindrome di Pena Shoker (artrogriposi multipla congenita), quest’ultimo nato morto.
Le condizioni cromosomiche sono 12 (7% sul totale; 14,8% dei casi associati), delle quali metà con atresia esofagea associata a fistola. Tali casi sono: 8 sindromi di Down (dei quali 5 con atresia esofagea senza fistola), 3 casi di trisomie 18 (tutte con atresia esofagea associata a fistola) e un caso di XXY (con atresia esofagea senza fistola).
Tutti i casi di anomalie cromosomiche sono state confermate con il cariotipo,
Tra i 5 casi di sindrome di Down con atresia esofagea senza fistola, 2 presentano anomalie anorettali.
Sex ratio
Dei 172 casi, 105 sono maschi e 67 femmine (M/F=1,56). Distinguendo secondo il tipo di atresia esofagea, la sex ratio nei casi di atresia esofagea con fistola è di 1,7 (63 M/37 F), mentre per i casi con atresia esofagea senza fistola è di 1,37 (42 M/30 F). Si osserva una predominanza maschile soprattutto per i casi di MCA con atresia esofagea senza fistola (15 M/7 F=2,14) e nelle condizioni note (prevalentemente costituite da VATER) con atresia esofagea associata a fistola tracheoesofagea (10 M/5 F=2). Tale dato è in accordo con i dati di letteratura che riportano una predominanza del sesso maschile in particolare nei casi associati.
Le condizioni cromosomiche presentano complessivamente una eguale distribuzione tra i due sessi
Gemellarità
Sono registrati 6 casi di gemelli con una frequenza del 3,5% sul totale. La frequenza di gemellarità è in accordo con i dati di letteratura. Tra essi 3 casi sono isolati e 3 sono MCA.
Peso alla nascita ed età gestazionale
Escludendo il caso di interruzione terapeutica, il peso medio alla nascita, è di circa 2560 g con un’età gestazionale media di 37 settimane. Le condizioni note con atresia esofagea presentano un peso medio (1960 g) e una media delle settimane di gestazione (34,7) più bassi.
Età materna
L’età media materna, escluse le condizioni cromosomiche,è di circa 30,1 aa, con una media di 29,7 per i casi con atresia esofagea (non significativo rispetto a popolazione generale), e di 30,5 aa per atresia esofagea associata a fistola (significativa rispetto a popolazione generale; p<0.001).
Ricordiamo che l’home page del sito IMER può essere consultata, per informazioni relative al registro, all’indirizzo http://www.unife.it/imer
Bibliografia
Brown AK, Roddam AW, Spitz L, Ward SJ (1999): Oesophageal atresia, related malformations, and medical problems: a family study. Am J Med Genet 85: 31-37.
Chittmittrapap S, Spitz L, Kiely M, Brereton RJ. (1989): Oesophageal atresia and associated anomalies. Arch Dis Child, 64, 364-368.
David TJ, O’Callaghan SE (1974): Twinning and oesophageal atresia. Arch Dis Child 49, 660-662.
David TJ, O’Callaghan SE (1975): Oesophageal atresia in the South West of England. J Med Genet, 12,1-11.
Depaepe A, Dolk H, Lechat MF and a EUROCAT working Group (1993): The epidemiology of tracheo-oesophageal fistula and oesophageal atresia in Europe. Arch Dis Child, 68: 743-748.
Di Gianantonio E, Schaefer C, Mastroiacovo P, Cournot MP, Benedicenti F, Reuvers M, Occupati B, Robert E, Bellemin B, Addis A, Arnon J, Clementi M (2001). Adverse effects of prenatal methimazole exposure. Teratology 64: 262-266.
Fraser C., Baird PA., Sadovnick AD. (1987): A comparison of incidence trends for esophageal atresia and tracheoesophageal fistula, and infectious disease. Teratology, 36: 363-369.
Harris J, Kallen B, Robert E (1995): Descriptive epidemiology of alimentary tract atresia. Teratology 52: 15-29.
Konstantinidou AE, Agapitos E, Korkolopoulou P, Davaris P. (2001): Tracheoesophageal malformation: pathogenetic evidence provided by two cases. Teratology 63: 11-14.
Lammer EJ, Cordero JF (1986): Exogenous sex hormone exposure and the risk for major malformations. JAMA, 255: 3128-3132.
Morini F, Cozzi DA, Ilari M, Casati A, Cozzi F (2001): Pattern of cardiovascular anomalies associated with esophageal atresia: support for a caudal pharyngeal arch neurocristopathy. Pediatr Res 50,5, 565-568.
Nora AH, Nora JJ. A (1975): Syndrome of multiple congenital anomalies associated with teratogenic exposure. Arch Environ Health, 30: 17-21.
Otten C, Migliazza L, Xia H, Rodriguez J, Diez-Pardo J, Tovar J (2000): Neural crest-derived defects in experimental esophageal atresia. Pediatr res. 47,2: 178-183.
Oxford J, Glasson M, Beasley S, Shi E. Myers N, Cass D. (2000): Oesophageal atresia in twins. Pediatr Surg Int 16: 541-545.
Possoegel AK, Diez-Pardo JA, Morales C, Tovar JA (1999). Notochord involvement in experimental esophageal atresia. Pediatr Surg Int, 15: 201-205.
Robert E, Mutchinick O. Mastroiacovo P, Knudsen LB, Dal Tveit AK, Castilla EE, Lancaster P, Kallen B, Cocchi G. (1993): An international collaborative study of the epidemiology of esophageal atresia or stenosis. Reprod Toxicol 7, 405-421.
Swipek A, Gregor V, Horacek J, Masatova D. (2002). Occurrence of congenital esophageal defects in the Czech Republic 1961-2000-incidence, prenatal diagnosis and prevalence according on maternal age. Ceska Gynekol 67, Suppl 1;: 29-32.
Sparey C, Robson SC.(2000): Oesophageal atresia. Prenat Diagn, 20, 251-253.
Seoud M, Nassar A, Usta I, Mansour M, Salti I, Younes K. (2003): Gastrointestinal malformations in two infants born to women with hyperthyroidism untreated in the first trimestre. Am J Perinatol 20(2): 59-62.
Squire EN (1977) :Seasonal occurrence of TE fistula. Birth Defects 12: 157-158.
Staey MV, De Bie S, Matton MT, De Roose J. (1984) : Familial congenital esophageal atresia. Hum Genet 66: 260-266.
Szendrey T, Danyl G, Czeizel A (1985) : Etiological study on isolated esophageal atresia. Hum Genet 70: 51-58.
Torfs CP, Milkovich L, Van den Berg B. (1981): The relationship between pregnancy tests and congenitsl anomalies. A prospective study. Am J Epidemiol. 113: 563-574.
Torfs CP, Curry CJR, Bateson TF. (1995): Population-based study of tracheoesophageal fistula and esophageal atresia. Teratology 52:220-232.
Van Heurn, Cheng W, De Vries B, Saing H, Jansen NJG, Kootstra G, Tam PKH. (2002): Anomalies associated with oesophageal atresia in Asians and Europeans. Pediatr Surg Int, 18: 241-243.
Warren J, Evans K, Carter CO. (1979): Offspring of patients with tracheo-oesophageal fistula. J Med Genet, 16, 338-340.