Gruppo IMER - Regione Emilia Romagna - NewsLetter 24
CRANIOSINOSTOSI
Calabrese O., Rivieri F., Calzolari E.
Con il termine craniosinostosi si intende la fusione prematura di una o più suture craniche.
Le suture craniche sono articolazioni di tipo fibroso che uniscono tra loro le ossa della volta cranica (costituita dalle ossa frontali, temporali, parietali e occipitali).
Normalmente le suture craniche si chiudono (cioè formano le sinostosi) solo in epoca postnatale; la sutura metopica (sutura della parte anteriore del cranio) si chiude attorno ai due anni, mentre le altre suture (sagittale, coronale e lambdoidea) si chiudono verso i 20 anni.
Nelle craniosinostosi, la saldatura delle suture può avvenire già in epoca prenatale o perinatale, oppure nel corso della prima infanzia, determinando una alterazione della forma del cranio .
Sviluppo del cranio e delle suture
Lo sviluppo del cranio nell’uomo è caratterizzato da tre fasi: formazione della ectomeninge, del tessuto cartilagineo e ossificazione. La formazione dell’ectomeninge, che deriva dal mesenchima delle creste neurali e dal mesoderma parassiale, inizia dalla 3° settimana di gestazione e dà luogo alle ossa della volta. Intorno alla 6-7 settimana, a livello della base cranica si ha la formazione di tessuto cartilagineo, che dà origine alle ossa del basicranio. Le cellule periferiche ai margini dei nuclei di ossificazione (dai quali si sviluppano le singole ossa del cranio), formano i cosidetti fronti osteogenici, che tendono ad approssimarsi dando luogo alle suture, costituite dai margini ossei giustapposti (come nelle suture della linea mediana, metopica e sagittale) o sovrapposti (come osservabile nelle altre suture). Successivamente si ha l’ossificazione diretta dell’ectomeninge, con formazione della volta cranica, e l’ossificazione del tessuto cartilagineo, che determina la base del cranio (Emery et al 2002).
La pervietà delle suture congiuntamente al rimodellamento delle ossa della volta, dato dall’equilibrio tra riassorbimento del tavolato interno e apposizione ossea all’esterno, consente l’espansione della scatola cranica sotto la spinta della crescita del cervello.
Il basicranio costituisce un campo di crescita entro il quale la componente nasomascellare e la mandibola si sviluppano; ne consegue che alterazioni del basicranio portano a precoci corrispondenti effetti sulle dimensioni, sulla configurazione e posizione delle varie parti maxillofacciali.
La forma della testa dipende da quali suture sono prematuramente saldate, dall’ordine e dallo stadio di sviluppo della formazione delle sinostosi. Più precocemente occorre la sinostosi, più importanti sono gli effetti sullo sviluppo del cranio.
Classificazione delle craniosinostosi
A seconda del coinvolgimento delle suture, le craniosinostosi possono essere semplici o complesse (una o più suture interessate).
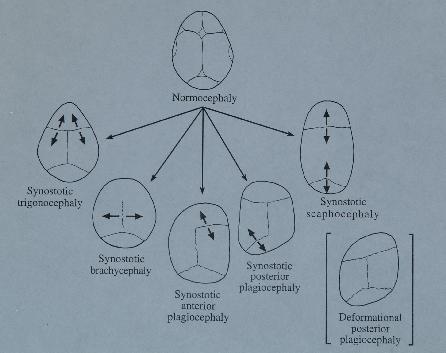
Riguardo alle sinostosi semplici, l’interessamento bilaterale della sutura coronale dà origine a brachicefalia con riduzione del diametro anteroposteriore del cranio. La prematura fusione della sutura sagittale dà luogo a dolicocefalia (con aumento del diametro anteroposteriore). Il coinvolgimento unilaterale della sutura coronale o lambdoidea causa plagiocefalia, con asimmetria della forma della testa. La fusione della metopica dà origine a trigonocefalia.
Nella figura 1 è illustrata la correlazione fra sutura interessata e conformazione cranica.
Le sinostosi complesse occorrono in circa il 5% dei casi di craniosinostosi non sindromiche (cioè non associate a definiti quadri clinici). Il coinvolgimento di 2 suture avviene in circa i 2/3 dei casi, e l’interessamento di più di due suture in un terzo dei casi. Spesso sono interessate suture contigue.
Più suture sono interessate, maggiore è il rischio di ritardo mentale.
Fig 1. Correlazione fra sutura interessata e conformazione cranica
Le craniosinostosi possono essere primarie, cioè non dovute a disordini associati, o secondarie, dovute a patologie metaboliche (ipertiroidismo, rachitismo, mucopolisaccaridosi), ematologiche (talassemia, anemia falciforme), malformazioni (oloprosencefalia, microcefalia, encefalocele), teratogeni (idantoina, retinoidi) o cause iatrogene (rapida decompressione per idrocefalia) (tab. 1).
|
Tabella 1. CONDIZIONI CON CRANIOSINOSTOSI SECONDARIE |
|
Disordini metabolici (ipertiroidismo, rachitismi, mucopilisaccaridosi) Disordini ematologici (talassemia, anemia falciforme) Malformazioni (oloprosencefalia, microcefalia, encefalocele, etc) Teratogeni: retinoidi Shunt per idrocefalia |
Le craniosinostosi occorrono spesso come anomalie isolate, ma possono associarsi ad altre malformazioni o patologie e possono costituire definiti quadri sindromici.
Le anomalie associate, soprattutto anomalie degli arti (84%), anomalie dell’orecchio (38%) e difetti cardiaci congeniti (23%), occorrono più frequentemente nelle sinostosi coronali che nelle sinostosi sagittali (Cohen et al 2000).
Sono note oltre 90 sindromi con craniosinostosi di cui circa la metà mostra modalità di ereditarietà mendeliana, a trasmissione autosomico dominante o recessiva, con ampia variabilità di espressione interfamiliare che intrafamiliare. Per diverse di queste sindromi, tra le quali Crouzon, Apert, Pfeiffer e Saethre-Chotzen, sono identificate le basi genetiche (vedi genetica delle craniosinostosi).
Sono state descritte molte condizioni cromosomiche che determinano craniosinostosi.
Epidemiologia delle craniosinostosi
Le craniosinostosi, nel loro complesso, sono tra le più comuni anomalie congenite con una prevalenza alla nascita di circa 1 su 2100-3000 nati nelle popolazioni europee, africane e asiatiche (Cohen et al 2000).
La casistica del registro IMER, dal 1978 al 2000, ha accertato 80 casi con craniosinostosi su un totale di 497.000 nati sorvegliati. La prevalenza alla nascita risulta di 0,16X1000 (cioè 1: 6250 nati). Tale dato indica una sottostima dovuta al periodo di rilevamento entro la prima settimana di vita.
La sinostosi sagittale è la più comune craniosinostosi e ha una prevalenza alla nascita di circa 1: 5000 nati. Oltre la metà dei casi occorre in forma isolata (cioè non associata ad altre anomalie congenite). La maggioranza dei casi sono sporadici (cioè senza ricorrenza familiare) con una predominanza del sesso maschile (rapporto M:F=3,5:1). Circa il 6% dei casi sono familiari e presentano, nella grande maggioranza dei casi, modalità di trasmissione autosomica dominante (Cohen et al 2000).
La sinostosi coronale ha una prevalenza alla nascita di circa 1: 10.000, e occorre isolata in circa la metà dei casi. La maggioranza dei casi sono sporadici, con una predominanza del sesso femminile (rapporto M:F = 1:2), e mostra una associazione con l’età paterna avanzata. Il 10-14% dei casi sono familiari con modalità di trasmissione autosomico dominante (Lajeunie et al. 1995, Cohen et al 2000).
La sinostosi metopica ha una prevalenza alla nascita di circa 1: 15.000, con una predominanza del sesso maschile (M:F= 3,3:1). Circa il 5,6% dei casi sono familiari (Cohen et al 2000).
La sinostosi lambdoidea presenta una prevalenza di 1:15.000 e predominanza del sesso maschile.
E’ importante distinguere nelle plagiocefalie posteriori, la presenza di una effettiva sinostosi lambdoidea dalle forme deformazionali, che risultano incrementate a seguito delle recenti prescrizioni pediatriche sulla postura nel sonno dei neonati, ove il cranio appare più comunemente di forma romboidale, anziché trapezoidale come osservabile nelle sinostosi (Fig 1) (Cohen et al 2000).
Fattori di rischio
Gli studi epidemiologici sulle craniosinostosi hanno evidenziato una predominanza del sesso maschile, in particolare per le sinostosi sagittale e lamdoidea (Singer et al 1999, Kallen 1999), mentre per le sinostosi coronali si evidenzia una predominanza del sesso femminile (Aldermann 1988, Singer et al 1999, Kallen 1999).
La prematurità (età gestazionale <37 settimane) e il basso peso alla nascita (<2500 g) sono riportati quali fattori di rischio (Alderman et al 1988, Singer 1999).
Non sono riportate differenze significative riguardo alla parità e alla razza (Alderman et al 1988, Kallen et al 1999).
Le craniosinostosi appaiono associate con l’incremento dell’età paterna (>40 aa) (Singer et al 1999, Kallen K 1999) in particolare per la sinostosi coronale. Anche l’età materna è stata riportata associata a tale tipo di craniosinostosi, (Alderman et al 1988, Singer et al 1999), seppur un altro studio (Kallen K 1999) non conferma tale dato.
Alcuni studi (Alderman et al 1988, Alderman et al 1994, Gardner et al 1998, Kallen K. 1999; Honein et al 2000) mostrano una associazione tra fumo materno in gravidanza e craniosinostosi isolate.
Genetica delle craniosinostosi
I geni dei recettori dei fattori di crescita dei fibroblasti (FGFRs) e il gene TWIST sono responsabili delle forme più comuni di craniosinostosi (Cohen et al 2000, Emery et al 2002).
I recettori dei fattori di crescita dei fibroblasti (FGFRs) sono costituiti da 4 diverse proteine, codificate da 4 geni situati su diversi cromosomi, cui si legano un numeroso gruppo (26 proteine) di fattori di crescita (FGFs).
La biologia dei recettori e dei fattori di crescita risulta molto complessa a causa della espressione spaziotemporale specifica, nel corso dello sviluppo embrionale, delle diverse isoforme dei recettori e dei numerosi ligandi.
Dalla più recente letteratura inerente i FGFRs si evidenzia:
Gli studi funzionali hanno evidenziato che tutte le mutazioni riconosciute comportano un guadagno di funzione della proteina recettoriale connessa a 1) una aumentata affinità di legame per i ligandi (come osservato per la mutazione Ser252Trp responsabile del 70% dei casi di S. di Apert), 2) a perdita di specificità del legame al ligando (come osservato, sempre nella S di Apert, per la mutazione P253R, osservata nella rimanente parte dei casi).
Il gene TWIST codifica per un fattore di trascrizione, inserito nella medesima via di sviluppo dei recettori FGFRs e agisce a monte di queste ultime. Le mutazioni in tale gene conducono ad una perdita di funzione della proteina e alcuni studi funzionali indicherebbero che una perdita di funzione di TWIST conduca ad una attivazione dei recettori FGFRs (cioè conduce a un guadagno di funzione dei FGFRs).
Per quanto riguarda le correlazioni genotipo-fenotipo (tra mutazione osservata e quadro clinico) la più consistente è osservabile nella sindrome di Apert ove due mutazioni (Ser252Trp e P253R), causanti sostituzioni aminoacidiche in due residui aminoacidici adiacenti, sono responsabili del 99% dei casi ed è possibile osservare una maggiore frequenza di schisi palatali nella prima e di sindattilia grave nella seconda (Emery et al 2002).
Nelle altre sindromi le correlazioni genotipo-fenotipo sono più difficili.
I dati molecolari hanno mostrato che le craniosinostosi finora considerate entità clinicamente distinte, possono mostrare mutazioni identiche o essere patologie alleliche (mutazioni diverse dello stesso gene).
E’ quindi auspicabile un nuovo sistema classificativo, che preveda una stretta collaborazione tra clinici e ricercatori per meglio definire i fattori eziologici (genetici/ambientali) capaci di influenzare il fenotipo
Tabella 2. Caratteristiche cliniche e genetiche di alcune craniosinostosi sindromiche
|
S. APERT |
S.CROUZON |
S. PFEIFFER |
S.SAETHRE-CHOTZEN |
S. MUENKE |
|
|
Prevalenza % sul totale |
1:60.000 4,5% |
1:60.000-25000 4,5% |
1:100.000 2,5% |
1:60.000 4,5% |
1:30.000 8% |
|
Sinostosi |
Coronale associato ad ampio difetto della linea mediana della volta |
Coronale, lambdoidea, saggitale |
Coronale (tipo I-III); cranio a trifoglio (tipo II) |
Coronale (incostante) |
Coronale uni/bilaterale, (incostante) |
|
Volto |
Fronte alta e prominente |
Proptosi (100%) |
Proptosi (Tipo III) |
Ptosi, crus auri prominente, |
Non specifico e incostante |
|
Estremità |
Sindattilia cutanea/ossea di almeno 3 dita |
Non coinvolte |
Pollice e alluci grandi |
sindattilia II-III dito a mani e piedi |
Non specifico e incostante (<50%) |
|
Alterazioni scheletriche |
Fusione vertebre cervicali C5-C6 |
Fusione vertebre cervicali C2-C3 |
Sinostosi/ anchilosi dei gomiti, fusione C2-C3 |
Fusione vertebre C2-C3 |
assente |
|
Anomalie associate |
Anomalie cerebrali, ritardo mentale (2/3 dei casi), anomalie gastrointestinali e urogenitali, acne |
Alta incidenza di complicanze SNC |
possibili complicanze SNC (tipo II- III) |
assenti |
assente |
|
Trasmissione-Occorrenza |
AD, sporadica per mutazioni de novo |
AD, 50% familiarità |
AD, sporadica (tipo II-III) Familiare (I) |
AD, familiare, alcuni casi sporadici |
AD, familiare |
|
Gene-mutazioni |
FGFR2 •S252W ( 2/3) •P253R (1/3) |
FGFR2, FGFR3
|
FGFR2 FGFR3 (P250R) FGFR1 (P252R) |
TWIST (80%) FGFR3 (P250R) |
14-52% FGFR3 (P250R) |
Desideriamo inoltre ricordarvi che il giorno 12 Aprile 2003 a Parma si terrà il:
XVIII Convegno Annuale IMER.
"Patologie scheletriche".
Il convegno verrà accreditato ECM
Bibliografia
Cohen MM Junior MacLean RE: Craniosinostosis II ed. Oxford University Press 2002
Emery DL, Connor JM, Pyeritz RE, Korf BR: Rimoin Emery and Rimoin’s Principles and Practice of Medical genetics Fourth edition. Churchill Livingstone 2002
Scriver CR, Beaudet AL, Sly WS, Valle D. The metabolic and molecular bases of inherited diseases. Eight edition. McGraw-Hill 2001
Lajeunie e et al. Genetic study of nonsyndromic coronal craniosynostosis. Am J Med Genet, 1995; 55: 500-504.
Singer S et al. Craniosynostosis in Western Australia. A population-based study. Am J Med Genet 1999; 83: 382-387.
Kallen K. Maternal smoking and craniosynostosis. Teratology 1999; 146-150.
Alderman BW et al. An epidemiologic study of craniosynostosis: Risk indicators for the occurrence of craniosynostosis in colorado. Am J Epidemiol 1988; 128: 431-438.
Alderman BW et al. Increased risk of craniosynostosis with maternal cigarette smoking during pregnancy. Teratology 1994; 50:13-18.
Gardner et al. Maternak exposure to prescription and non- prescription pharmaceuticals or drugs of abuse and risk of craniosynostosis. Int J Epidemiol 1998 27:64-67
Honein MA et al. Further evidence of an association between maternal smoking and craniosynostosis. Teratology 2000; 62: 145-149.
Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002 Jun 15;16(12):1446-65
Kan SH et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002 Feb;70(2):472-86.