 FIG.1
FIG.1
IL GLAUCOMA CONGENITO PRIMARIO
DEFINIZIONE
Il glaucoma è una patologia oftalmologica caratterizzata da una neuropatia ottica a decorso acuto, subacuto , o cronico-progressivo che esita nella perdita di cellule ganglionari retiniche e nella conseguente atrofizzazione del nervo ottico.
In rapporto all’età di esordio delle manifestazioni cliniche è possibile distinguere un glaucoma infantile ( definito anche "congenito " o "malformativo"), un glaucoma dell’adolescente ed un glaucoma dell’adulto.
EMBRIOGENESI
Il substrato patogenetico comune di tutti i glaucomi congeniti è rappresentato da una alterazione della trabeculogenesi (trabeculodisgenesi) , il processo embriogenetico che conduce alla genesi del trabecolato sclero-corneale , la struttura anatomo-funzionale a livello della quale si effettua il deflusso dell’umore acqueo. La vescicola lenticolare ( il cristallino embrionale) nasce da una invaginazione della superficie ectodermica dalla quale si separa completamente in prossimità della VI settimana gestazionale , quando il calice ottico (la retina embrionale) di origine neuroectodermica raggiunge la periferia della vescicola: una massa triangolare di cellule derivate dalla cresta neurale si distaccano dal calice ottico per raggiungere l’ectoderma dell’epitelio corneale dando origine a stroma ed endotelio corneale, trabecolato, stroma irideo.
Tripathy (1) ha mostrato che le cellule del trabecolato contengono enolasi neuro-specifiche rivelandone l’origine embriologica dalla cresta neurale.
Dal momento che la trabeculodisgenesi costituisce una alterazione delle fasi di migrazione, cavitazione, ed atrofizzazione della massa cellulare originante dalla cresta neurale nell’ambito del processo morfogenetico della camera anteriore dell’occhio il glaucoma congenito può essere definito sotto il profilo embriopatogenetico una neurocrestopatia focale .
PATOGENESI
Nell’occhio affetto da glaucoma congenito l’umore acqueo normalmente prodotto dall’epitelio del corpo ciliare attraversa il forame pupillare ma incontra elevate resistenze idrodinamiche a livello dell’angolo camerulare nell’attraversare le maglie di un trabecolato sclerocorneale malformato e collassato , il che si traduce in un incremento della pressione intraoculare ; nei primi 18 mesi di vita, quando il collageno corneale e sclerale risulta ancora elastico, una pressione intraoculare elevata può deformare la cornea , distendere la sclera e determinare un aumento complessivo delle dimensioni oculari .
ASPETTI CLINICI
All’esame clinico la cornea appare ingrandita (megalocornea) con un diametro di 14-15 mm invece dei normali 10-11 mm (2); il diametro corneale dei prematuri risulta essere circa 8 mm (3). La membrana del Descemet , la base su cui poggiano le cellule dell’endotelio corneale , viene spesso lacerata dalla spinta pressoria per cui diventano visibili strie multiple (strie di Haab) orientate in senso orizzontale o curvilineo, concentriche al limbus corneale. L’ipertono oculare induce una miopia di tipo assile: il glaucoma congenito viene denominato anche buftalmo (occhio di bue, "ox eye"), termine che indica la coesistenza di miopia assile e di megalocornea , cioè un incremento complessivo di tutti i diametri oculari.
In tutti i neonati con megalocornea che rivelano irritabilità e fotofobia bisogna sospettare un glaucoma congenito ed indirizzare i genitori verso una diagnosi specialistica che consentirà , per mezzo di una gonioscopia l’osservazione del trabecolato malformato (membrana di Barkan) , per mezzo di una tonometria sotto narcosi la rilevazione dell’ipertensione oculare (ipertono), per mezzo di una fundoscopia l’accertamento del danno anatomico a livello della testa del nervo ottico (escavazione papillare), per mezzo di un esame biomicroscopico della cornea l’individuazione del danno alla membrana del Descemet (strie di Haab).
CLASSIFICAZIONE
Nel glaucoma congenito primario (GCP idiopatico) l’unica alterazione patologica presente nell’individuo è rappresentato dalla trabeculodisgenesi. Nel glaucoma congenito secondario (GCS) la condizione patologica glaucomatosa si iscrive nel contesto di quadri clinici più complessi:
Si definisce "sindrome angolare" una sindrome oculare in cui il difetto embriogenetico non coinvolge esclusivamente la trabeculogenesi (GCP) ma si estende anche all’iride, all’endotelio corneale ed all’intero angolo camerulare (iridogoniodisgenesi).
DATI EPIDEMIOLOGICI IN EMILIA-ROMAGNA
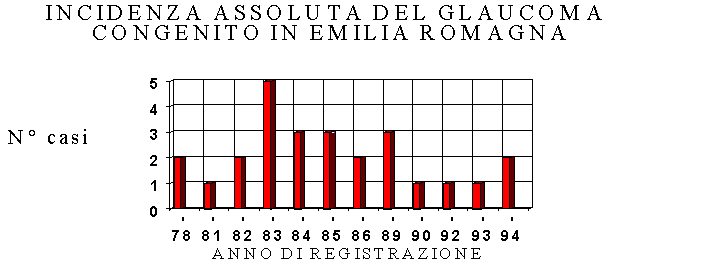
Dal 1978 al 1994 i casi di glaucoma congeniti registrati dall’IMER risultano pari a 26 (fig.1), con una netta prevalenza di glaucoma congenito primario [81%(fig.2)]. In Emilia-Romagna la prevalenza alla nascita risulta pari a 0.75/10.000; negli altri registri italiani tale valore è compreso tra 0,13 (Firenze) e 0,43 (Umbria) per 10.000) (4) . La prevalenza media alla nascita registrata nelle casistiche internazionali risulta essere di 1/10.000 circa (5).
FIG.1
 FIG.2
FIG.2
Sono stati registrati (fig.2) 14 casi di GCP isolato , 1 caso di GCP associato a S.di Klinefelter, 1 caso di GCP associato a S. di Noonan, 5 casi di GCP associato a sindromi polimalformative (S.Veil-Marchesani, Artrogriposi, Difetto interventricolare, Macrocefalia con microrinia, Micrognazia con naso adunco e clinodattilia).
Sono stati registrati 3 casi di sindrome di Axenfeld-Rieger e due casi di Aniridia (GCS:sindromi angolari focali).
Nei glaucomi congeniti primari si riscontra familiarità positiva nel 14% dei casi (3/21) e insorgenza sporadica nel residuo 86% di casi [le casistiche Europee riportano 7 casi sporadici su 8 (87.5%)]. L’interessamento è bilaterale nel 96% dei casi.
Due casi dei tre di S. di Axenfeld-Rieger presentano associazione con DIA (difetto interatriale).
ASPETTI GENETICI
Il glaucoma congenito primario (GCP) è una oftalmopatia su base genetica che risulta responsabile dello 0.01-0.04% dei casi di cecità totale (6). E’ stata ipotizzata una modalità di trasmissione autosomico recessiva con penetranza variabile (7), sebbene studi più recenti abbiano rilevato nella maggior parte dei casi un modello di ereditarietà poligenico e multifattoriale (8,9).
Allorquando il GCP si è manifestato nei gemelli monozigoti sono sempre stati affetti tutti i gemelli (10): è da segnalare però un caso in cui, pur sussistendo la gemellarità monozigotica, l’altro gemello è risultato indenne da patologia , rivelando nella fattispecie la presenza di elementi condizionati ambientali (11). Anomalie cromosomiche sono state frequentemente riportate associate al glaucoma congenito sebbene solo raramente si accompagnino alla classica sindrome del glaucoma congenito primario [trisomia 2q, monosomia 9p (12)]: qualora sussistano anomalie morfostrutturali cromosomiche evidenziabili con l’analisi citogenetica (es. inversione pericentrica del chr.11) il fenotipo clinico è quasi sempre caratterizzato da dismorfismi ed alterazioni patologiche multisistemiche (Schinzel, 1994).
I casi a trasmissione autosomico recessiva rivelano un fenotipo clinico di maggiore gravità che assume le seguenti caratteristiche( Genkik, 1989):
E’ evidente come anche nel glaucoma congenito primario sia presente eterogeneità genica, purtuttavia studi di linkage hanno circoscritto un locus specifico (GLC3A) sul braccio corto del cromosoma 2 (regione 2p21). Sarfarazi (6) ha riscontrato tale associazione nel 70 % della sua casistica (Connecticut,USA) . Analisi di linkage multiplo con 14 markers del braccio corto del cromosoma 2 hanno consentito di costruire la seguente mappa in direzione telomero-centromero:
TEL-D2S405-D2S367-(D2S1788-D2S1325)-[(GL3A++++, D2S177)/(D2S1346/D2S1348)]- D2S1356-D2S119-D2S1761-D2S1248-D2S1352-D2S406-D2S441-CEN
Dei sette geni che mappano alla regione 2p21 , CAD, CALM2, LHGCR, risultano centromerici rispetto a D2S119 e devono essere esclusi come geni candidati per il locus GLC3A : gli altri 4 geni [PRKR (P68 chinasi inducibile dall’interferone), TIK (serina-treonina chinasi), SOS1 (gene omologo della drosophila) , SPTBN1 ( forma extraeritrocitaria della beta spettrina)] non possono essere esclusi come geni candidati a costituire il substrato del fenotipo clinico del GLCP.
Per la sintenia esistente tra la regione 2p21 ed il chr. murino 11 (SPTBN1) ed il chr. murino 17 (TIK e SOS1) il locus omologo al GLC3A umano dovrebbe essere localizzato e ricercato sui chr.11 e 17 del topo.
Counselling genetico
I casi sporadici rappresentano più dell’ 85% nelle casistiche europee.
Il rischio di avere un altro figlio affetto dopo il primo è valutabile empiricamente intorno al 10% : dopo due figli affetti la trasmissione si considera autosomico-recessiva ed il rischio si eleva al 25%.
Considerando che nella popolazione di riferimento sone presenti quote variabili di casi con trasmissione A.R. e poligenica multifattoriale si può considerare un rischio generico empirico del 5% per i figli di individui affetti (13).
Bibliografia